Category: Publications
-
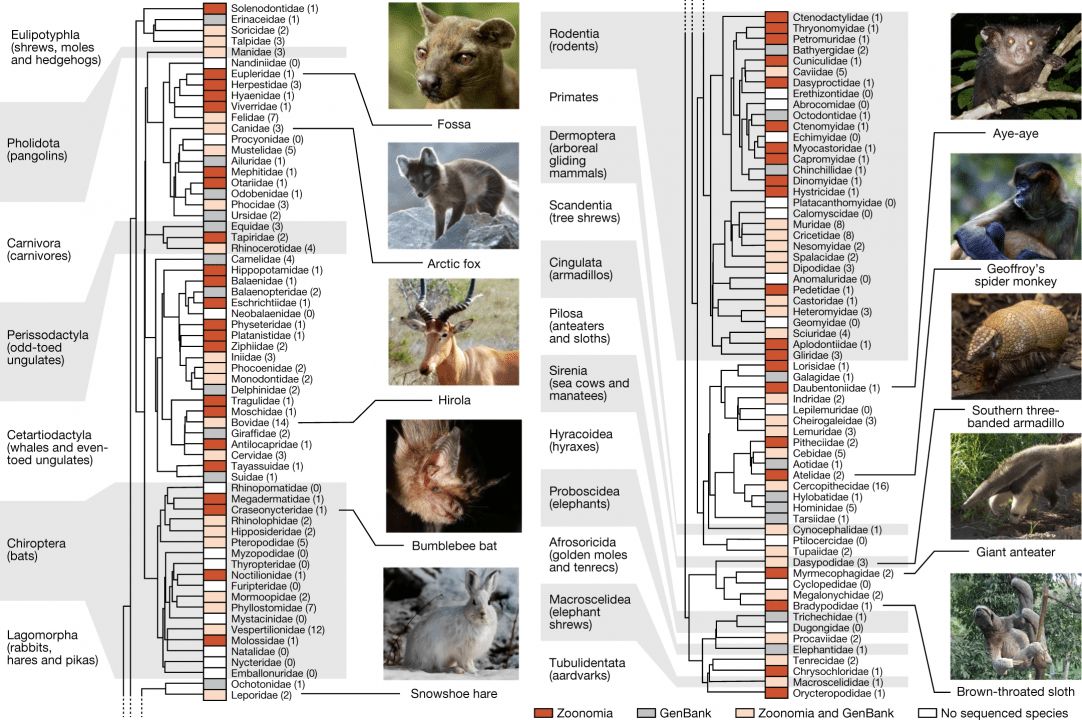
A comparative genomics multitool for scientific discovery and conservation
Zoonomia Consortium Abstract The Zoonomia Project is investigating the genomics of shared and specialized traits in eutherian mammals. Here we provide genome assemblies for 131 species, of which all but 9 are previously uncharacterized, and describe a whole-genome alignment of 240 species of considerable phylogenetic diversity, comprising representatives from more than 80% of mammalian families.…
-

Dense sampling of bird diversity increases power of comparative genomics
Shaohong Feng, Josefin Stiller, […] Guojie Zhang Abstract Whole-genome sequencing projects are increasingly populating the tree of life and characterizing biodiversity. Sparse taxon sampling has previously been proposed to confound phylogenetic inference, and captures only a fraction of the genomic diversity. Here we report a substantial step towards the dense representation of avian phylogenetic and…
-

Progressive Cactus is a multiple-genome aligner for the thousand-genome era
Joel Armstrong, Glenn Hickey, Mark Diekhans, Ian T. Fiddes, Adam M. Novak, Alden Deran, Qi Fang, Duo Xie, Shaohong Feng, Josefin Stiller, Diane Genereux, Jeremy Johnson, Voichita Dana Marinescu, Jessica Alföldi, Robert S. Harris, Kerstin Lindblad-Toh, David Haussler, Elinor Karlsson, Erich D. Jarvis, Guojie Zhang, and Benedict Paten Abstract New genome assemblies have been arriving…
-

AMELIE speeds Mendelian diagnosis by matching patient phenotype and genotype to primary literature
Finding a gene in the stacks Genetic disease diagnosis can be time-consuming because of the extensive literature searching required. To speed this process, Birgmeier et al. developed AMELIE (Automatic Mendelian Literature Evaluation), an end-to-end machine learning approach with web interface that finds relevant literature supporting the disease causality of genetic variants and their association with different…
-
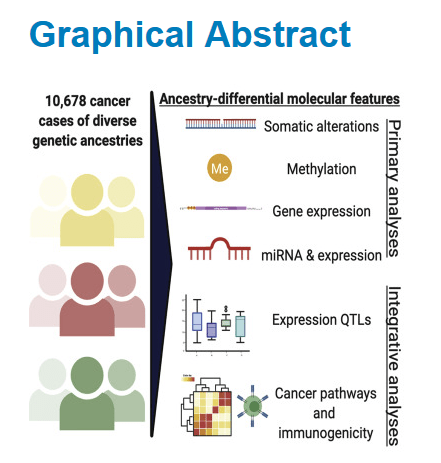
Comprehensive Analysis of Genetic Ancestry and Its Molecular Correlates in Cancer
Highlights This large analysis identified ancestry correlates in cancer Ancestry-associated artifacts and confounders were identified Ancestry effects are profoundly tissue specific Rates of FBXW7, VHL, and PBRM1 mutations and immune activity vary by ancestry Summary We evaluated ancestry effects on mutation rates, DNA methylation, and mRNA and miRNA expression among 10,678 patients across 33 cancer…
-

Domain-Specific Hardware Accelerators Communications of the ACM
From the simple embedded processor in your washing machine to powerful processors in data center servers, most computing today takes place on general-purpose programmable processors or CPUs. CPUs are attractive because they are easy to program and because large code bases exist for them. The programmability of CPUs stems from their execution of sequences of…
-
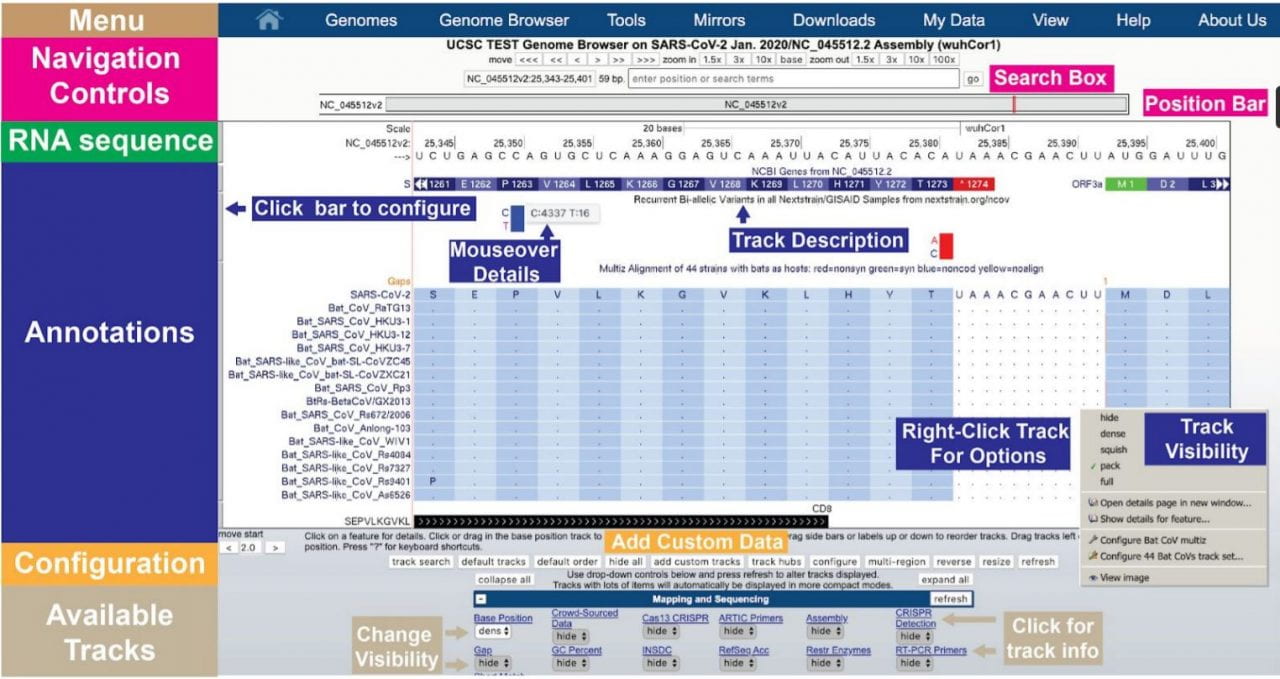
The UCSC SARS-CoV-2 Genome Browser
The UCSC Genome Browser features a Coronavirus Browser to help researchers visualize the coronavirus genome that causes COVID-19. Refer to the tutorial and video here: http://bit.ly/3qzPyQd. UCSC’s coronavirus browser maps gene and protein alignments, global variants, and even the genomes of variants from other species like bats and pangolins. It draws on sequencing data from around the world,…
-
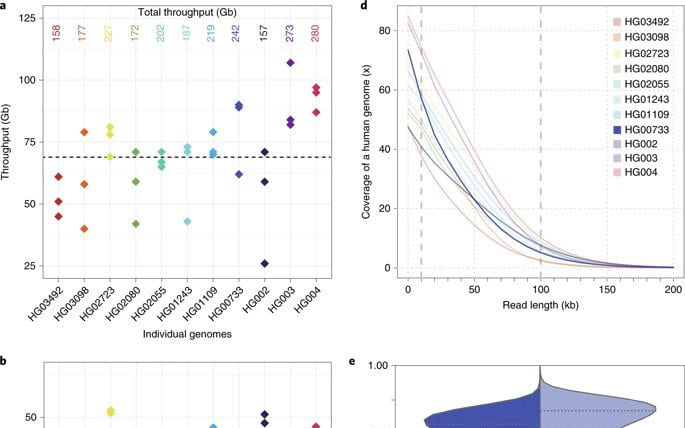
Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes
Abstract De novo assembly of a human genome using nanopore long-read sequences has been reported, but it used more than 150,000 CPU hours and weeks of wall-clock time. To enable rapid human genome assembly, we present Shasta, a de novo long-read assembler, and polishing algorithms named MarginPolish and HELEN. Using a single PromethION nanopore sequencer and…
-
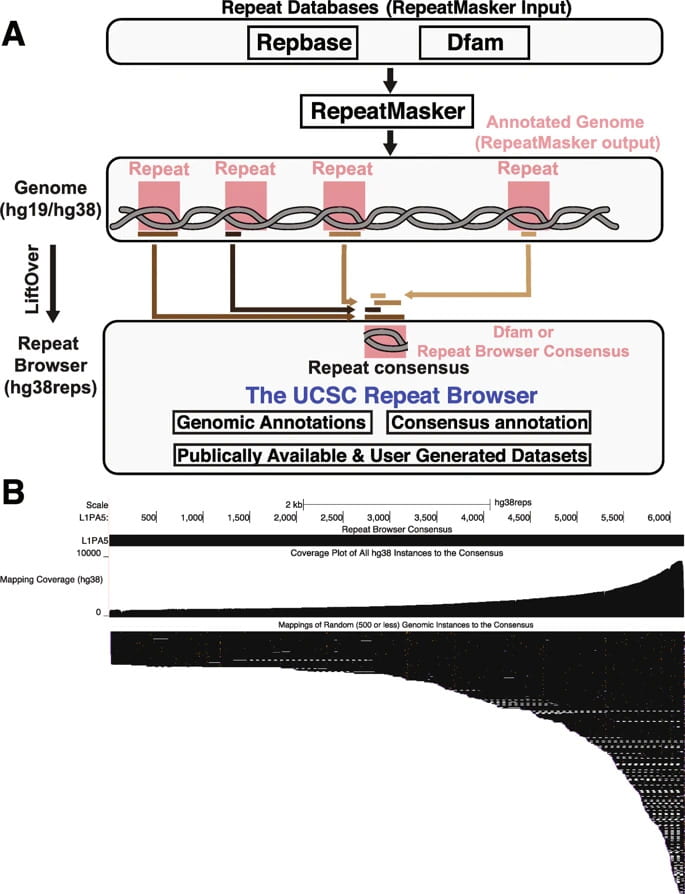
The UCSC Repeat Browser allows discovery and visualization of evolutionary conflict across repeat families
Mobile DNA | 31 March, 2020 Jason D. Fernandes, Armando Zamudio-Hurtado, W. James Kent, David Haussler, Sofie R. Salama, Maximilian Haeussler Abstract Background Nearly half the human genome consists of repeat elements, most of which are retrotransposons, and many of these sequences play important biological roles. However repeat elements pose several unique challenges to current bioinformatic analyses and visualization tools, as short repeat sequences can map to…
-

The case of an arctic wild ass highlights the utility of ancient DNA for validating problematic identifications in museum collections
Alisa O. Vershinina, Joshua D. Kapp, Gennady F. Baryshnikov, Beth Shapiro Molecular Ecology Resources | December 23, 2019 Abstract Museum collections are essential for reconstructing and understanding past biodiversity. Many museum specimens are, however, challenging to identify. Museum samples may be incomplete, have an unusual morphology, or represent juvenile individuals, all of which complicate accurate…
-
Highly Multiplexed Single-Cell Full-Length cDNA Sequencing of human immune cells with 10X Genomics and R2C2
Roger Volden, Christopher Vollmers bioRxiv | January 11, 2020 Abstract Single cell transcriptome analysis elucidates facets of cell biology that have been previously out of reach. However, the high-throughput analysis of thousands of single cell transcriptomes has been limited by sample preparation and sequencing technology. High-throughput single cell analysis today is facilitated by protocols like the 10X Genomics platform…
-

Off Earth Identification of Bacterial Populations Using 16S rDNA Nanopore Sequencing
Article | MDPI | 9 January 2020 Aaron S. Burton, Sarah E. Stahl, Kristen K. John, Miten Jain, Sissel Juul, Daniel J. Turner, Eoghan D. Harrington, David Stoddart, Benedict Paten, Mark Akeson, and Sarah L. Castro-Wallace Abstract The MinION sequencer has made in situ sequencing feasible in remote locations. Following our initial demonstration of its high performance off planet with Earth-prepared samples, we developed and tested an end-to-end,…