- 1 UC Santa Cruz Genomics Institute, Santa Cruz, CA, USA
- 2 University of Amsterdam, Swammerdam Institute for Life Sciences, Amsterdam, the Netherlands
- 3 Molecular, Cell and Developmental Biology Department, UC Santa Cruz, Santa Cruz, CA, USA
- 4 Department of Computer Science, Stanford University, Stanford, CA, USA
- 5 Howard Hughes Medical Institute, UC Santa Cruz, Santa Cruz, CA, USA
- 6 Department of Neurology and the Eli and Edythe Broad Center for Regeneration Medicine and Stem Cell Research at the University of California, San Francisco, San Francisco, CA, USA
- 7 Department of Genome Sciences, University of Washington School of Medicine, Seattle, WA, USA
- 8 Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA, USA
- 9 Department of Neurology, Harvard Medical School, Boston, MA, USA
- 10 Program in Medical and Population Genetics and Stanley Center for Psychiatric Research, Broad Institute, Cambridge, MA, USA
- 11 Service de génétique médicale, Lausanne, Switzerland
- 12 Department of Cytogenetics, Northern Genetics Service, Institute of Genetic Medicine, Newcastle upon Tyne, UK
- 13 Howard Hughes Medical Institute, University of Washington, Seattle, WA, USA
Highlights
• NOTCH2NLA,B,C expressed in human fetal brain radial glia stem cells, arose 0.5–4 mya
• These genes encode secreted, Notch-related proteins that enhance Notch signaling
• Overexpression of NOTCH2NL delays neuronal differentiation, while deletion accelerates it
• NOTCH2NLA and NOTCH2NLB serve as breakpoints in 1q21.1 deletion/duplication syndrome
Summary
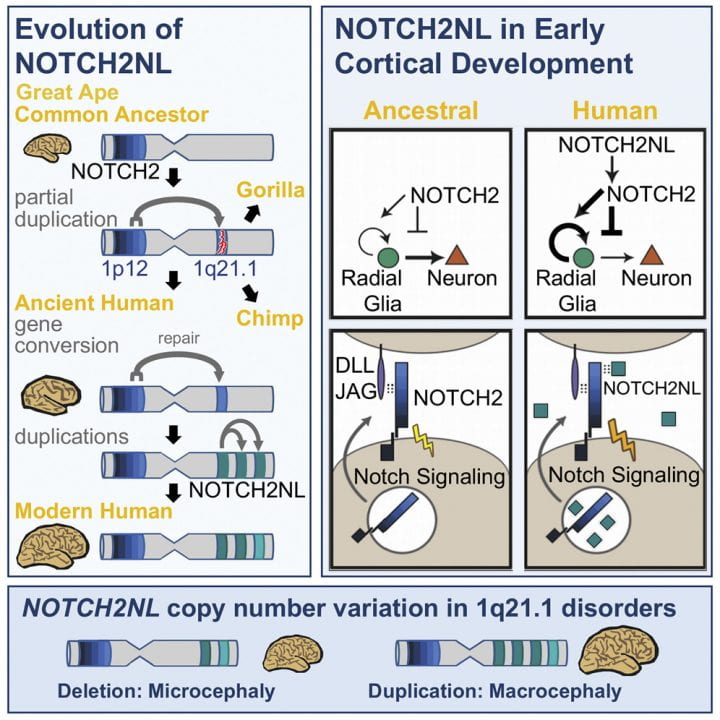
Genetic changes causing brain size expansion in human evolution have remained elusive. Notch signaling is essential for radial glia stem cell proliferation and is a determinant of neuronal number in the mammalian cortex. We find that three paralogs of human-specific NOTCH2NL are highly expressed in radial glia. Functional analysis reveals that different alleles of NOTCH2NL have varying potencies to enhance Notch signaling by interacting directly with NOTCH receptors. Consistent with a role in Notch signaling, NOTCH2NL ectopic expression delays differentiation of neuronal progenitors, while deletion accelerates differentiation into cortical neurons. Furthermore, NOTCH2NL genes provide the breakpoints in 1q21.1 distal deletion/duplication syndrome, where duplications are associated with macrocephaly and autism and deletions with microcephaly and schizophrenia. Thus, the emergence of human-specific NOTCH2NL genes may have contributed to the rapid evolution of the larger human neocortex, accompanied by loss of genomic stability at the 1q21.1 locus and resulting recurrent neurodevelopmental disorders.
Introduction
Human brains are characterized by a large neocortex that forms the substrate for the development of human-specific cognitive functions (Molnár et al., 2006), but evolutionary changes to our genome underlying this increase in size and complexity are poorly understood (Varki et al., 2008). Structural genomic variants account for 80% of human-specific base pairs (Cheng et al., 2005). Of particular interest are loci where segmental duplications have created entirely new human-specific gene paralogs associated with cortical development, such as SRGAP2C, ARHGAP11B, and TBC1D3 (reviewed inDennis and Eichler [2016]). Human-specific duplicated genes are often located within segmental duplications that mediate recurrent rearrangements associated with human disease ( Stankiewicz and Lupski, 2010 ; Popesco et al., 2006). One such region lies on human chromosome band 1q21, which was subject to a large pericentric inversion involving considerable gene loss and duplication during human evolution (Szamalek et al., 2006). The 1q21 locus contains a disproportionate number of human-specific genes (O’Bleness et al., 2012) and also contains the 1q21.1 distal deletion and duplication syndromes (henceforth referred to as 1q21.1 distal deletion/duplication syndrome) interval ( Mefford et al., 2008 ; Brunetti-Pierri et al., 2008). De novo deletion of one copy frequently leads to brain size reduction (microcephaly) and duplication to brain size increase (macrocephaly), among other symptoms.
1q21.1 was incorrectly assembled in the human reference genome until the most recent version, GRCh38, (Steinberg et al., 2014). In the revised assembly, this locus harbors three human-specific NOTCH2-derived genes we call NOTCH2NLA, NOTCH2NLB, and NOTCH2NLC, which are highly expressed in human cortical progenitors but were never considered candidates for the 1q21.1 syndromes. Notch signaling is central to brain development, determining the timing and duration of neuronal progenitor proliferation and neuronal differentiation ( Hansen et al., 2010). It is active in outer radial glia (oRG), a cell type hypothesized to generate the majority of primate cortical neurons and to contribute to human-specific cortical expansion (Lui et al., 2011). Through functional, genomic, and evolutionary analysis, we find evidence that with the creation of the modern forms of NOTCH2NL genes in the last few million years after divergence from chimpanzees, humans gained new, secreted NOTCH-like proteins that can enhance Notch signaling and may prolong cortical neurogenesis by delaying differentiation of neural progenitors. Our data suggest that the emergence of NOTCH2NL genes in humans may have contributed to the increase in size and complexity of the human neocortex at the expense of susceptibility to 1q21.1 distal duplication/deletion syndrome.